Adding columns of text
Use the col.left and col.right arguments to add columns of text either side of each panel. Use col.left.heading and col.right.heading to customise the column headings.
resultsA <- dplyr::filter(ckbplotr_forest_data, name == "A")
resultsB <- dplyr::filter(ckbplotr_forest_data, name == "B")
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Triglycerides concentration",
"Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.headings = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
col.heading.space = 1.5)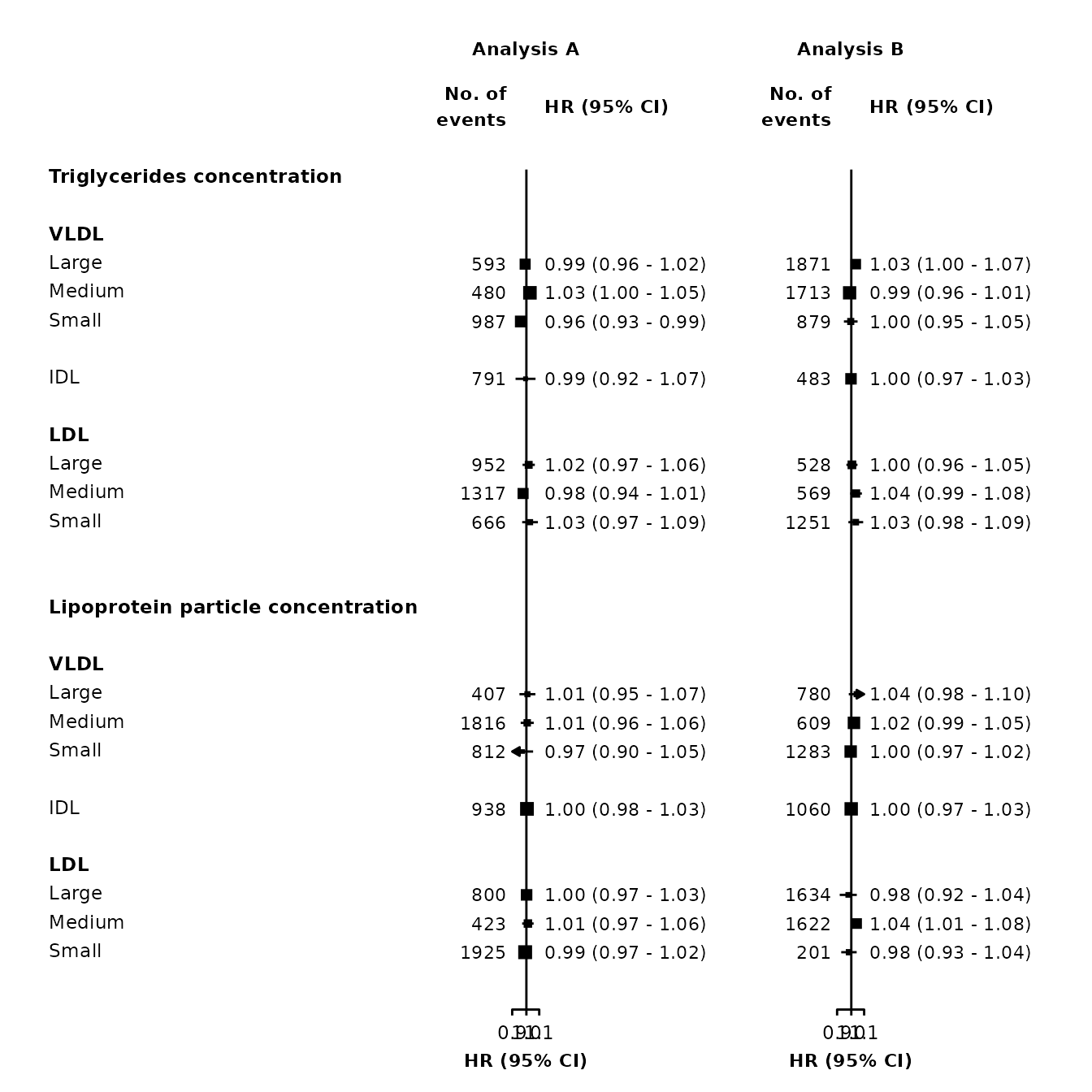
Multiple columns can be added by specifying vectors for col.left, col.right, col.left.heading and col.right.heading.
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Triglycerides concentration",
"Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.headings = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("nb", "n"),
col.left.heading = c("Some other\nnumbers", "No. of\nevents"),
col.right = "n",
col.right.heading = c("HR", "N"),
col.heading.space = 1.5)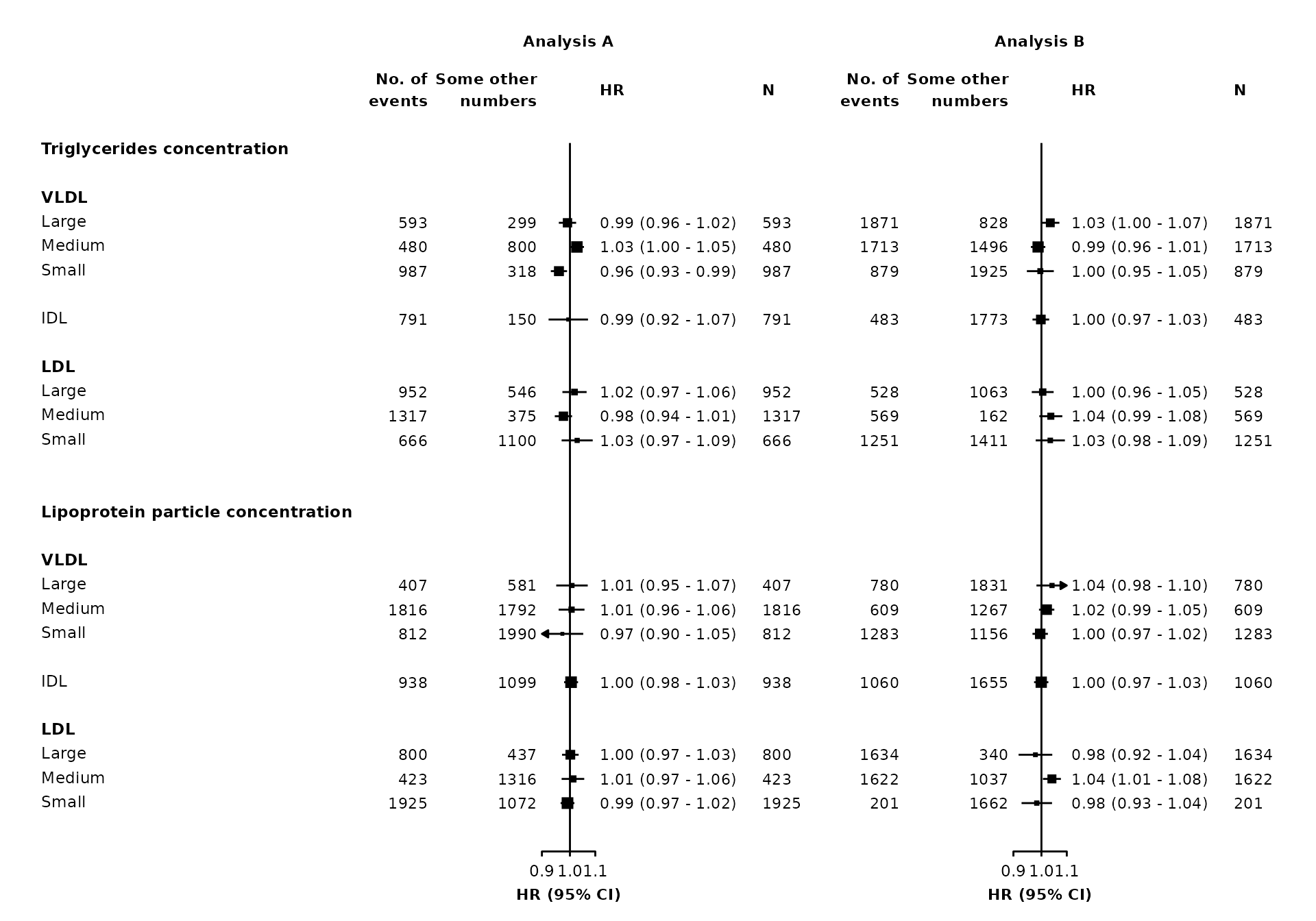
The col.left.hjust and col.right.hjust arguments set the horizontal justification of the columns (0 = left, 0.5 = center , 1 = right).
Spacing
Plot colour
The colour used for the plot can be changed with the plotcolour argument. This can be useful to create plots that fit a colour scheme (or use a dark grey for less contrast when viewing on a screen or projector). See the next section for details on customising the colour(s) of point and confidence interval lines.
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Triglycerides concentration",
"Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.headings = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
plotcolour = "navyblue")
Setting colours and shapes, bold text and diamonds
The shape and fill colour of points, colour of points and confidence interval lines, bold text, and which estimates/CIs should be plotted as diamonds can be set overall or on a per-point basis. This is done by setting arguments shape, colour, fill, ciunder, col.bold, and col.diamond to appropriate values, or to the name of a column containing values for each point.
The argument/columns, what they control, and the type:
| argument | controls | type |
|---|---|---|
| shape | plotting character for points | integer |
| colour | colour of points and lines | character |
| fill | fill colour of points | character |
| ciunder | if the CI line should be plotted before the point | logical |
| col.bold | if text is bold | logical |
| col.diamond | if a diamond should be plotted | logical |
Note that col.bold, and col.diamond must be column names in the supplied data frames, while the others can be fixed values or column names.
Using values
forestplot1 <- make_forest_plot(panels = list(resultsA),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Triglycerides concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A"),
blankrows = c(0, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
shape = 16,
colour = "red",
cicolour = "black",
ciunder = TRUE)
Using column names
resultsA[9,"shape"] <- 16
resultsA[10, "bold"] <- TRUE
resultsA[11, "colour"] <- "red"
resultsA[12, "diamond"] <- TRUE
resultsA[13, "ciunder"] <- TRUE
resultsA[13, "shape"] <- 22
resultsA[13, "fill"] <- "white"
forestplot1 <- make_forest_plot(panels = list(resultsA),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Triglycerides concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A"),
blankrows = c(0, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
shape = "shape",
colour = "colour",
col.bold = "bold",
col.diamond = "diamond",
ciunder = "ciunder")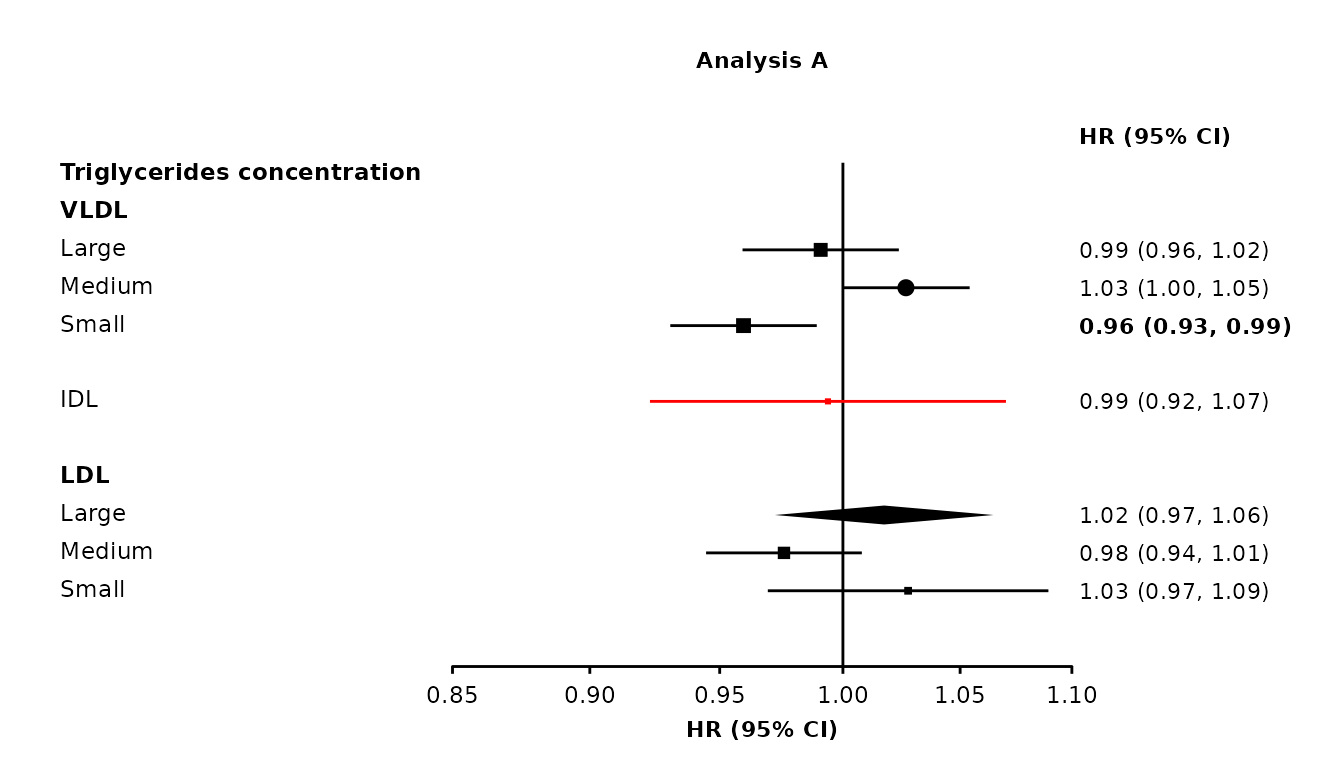
If a parameter is not set, then default values are used for these aesthetics. If a parameter is set, then every data frame provided in cols must contain a column with that name.
Diamond shortcut
As an alternative to using col.diamond, provide a character vector in the diamond argument. In rows with these key values, estimates and CIs will be plotted using a diamond. (If a list is supplied, only the first element will be used.)
forestplot1 <- make_forest_plot(panels = list(resultsA),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Triglycerides concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A"),
ci.delim = " - ",
blankrows = c(0, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
diamond = c("nmr_l_ldl_tg", "nmr_m_ldl_tg"))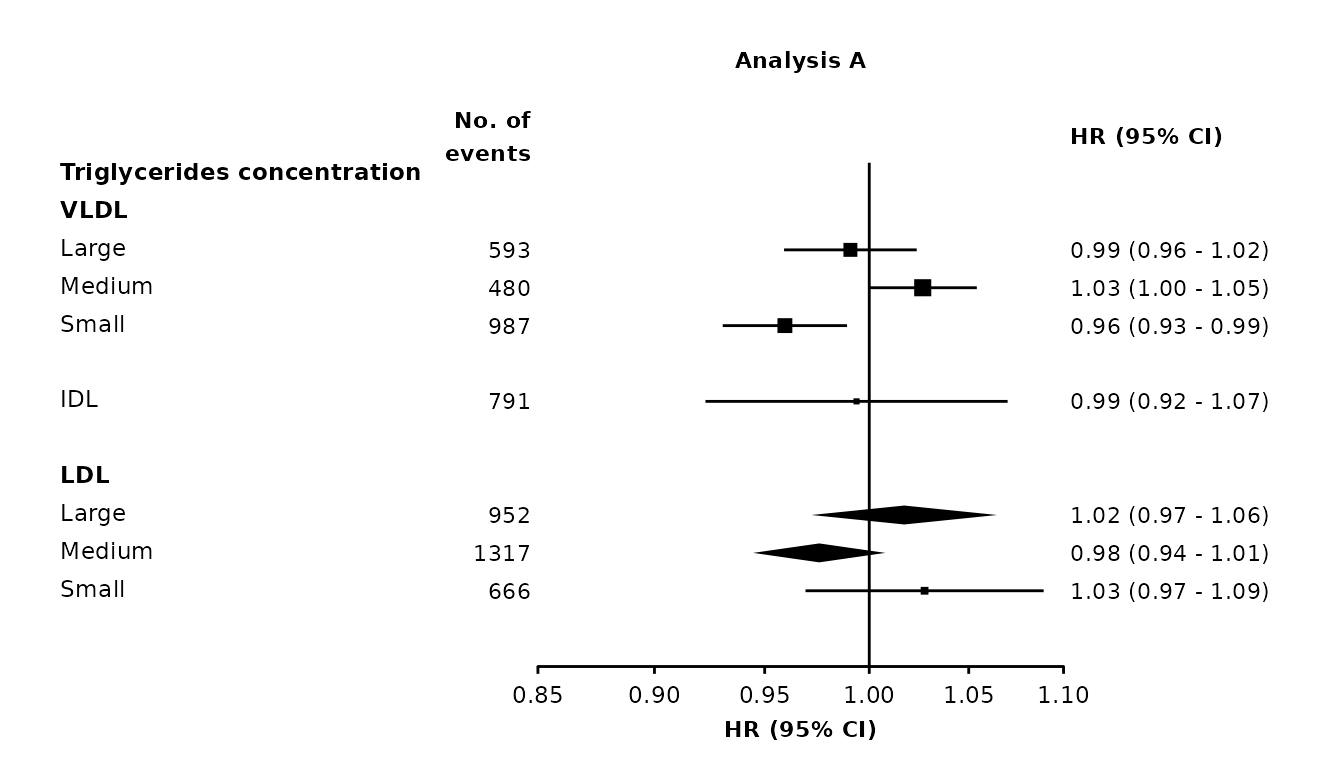
Adding heterogeneity and trend test results and other text
The addtext argument can be used to add results of heterogeneity or trend tests, or some other text, in the text column of estimates and CIs.
The automatic positioning of columns and spacing of panels does not take into account this additional text, so you may need to use the right.space and col.right.pos arguments for a satisfactory layout.
resultsA_extra
#> variable het_dof het_stat het_p trend_stat trend_p
#> 1 nmr_s_ldl_p 2 12 =0.22 <NA> <NA>
#> 2 nmr_s_vldl_p <NA> <NA> <NA> 7 =0.31
resultsB_extra
#> variable het_dof het_stat het_p trend_stat trend_p
#> 1 nmr_s_ldl_p 2 14 =0.32 <NA> <NA>
#> 2 nmr_s_vldl_p <NA> <NA> <NA> 7 =0.83
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration",
"Triglycerides concentration"),
exponentiate = TRUE,
panel.headings = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 0, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
col.heading.space = 1.5,
addtext = list(resultsA_extra, resultsB_extra),
right.space = unit(35, "mm"))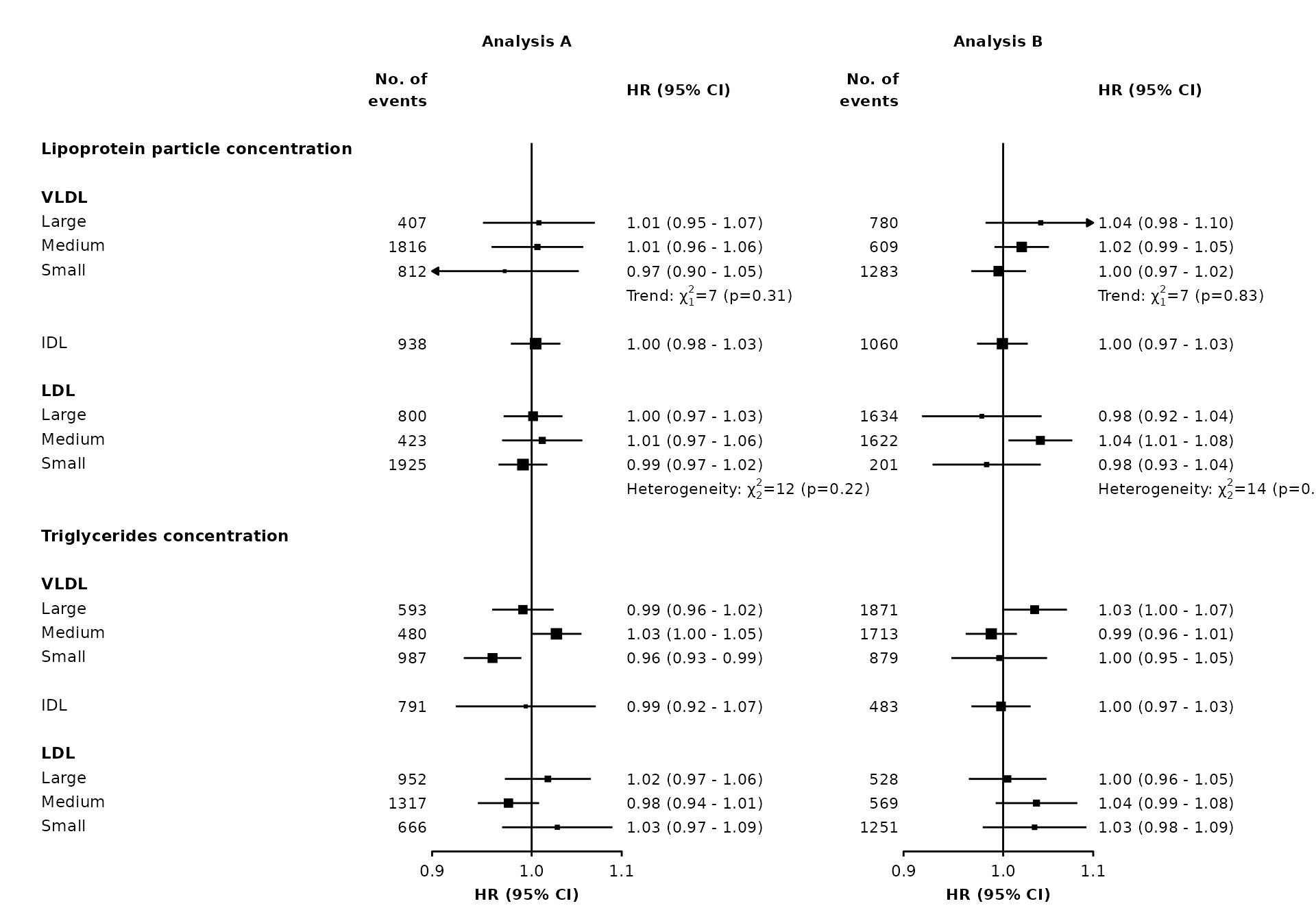
To add multiple tests results and/or text under the same row, use separate in the addtext data frames:
resultsA_extra
#> variable het_dof het_stat het_p trend_stat trend_p
#> 1 nmr_s_ldl_p 2 12 =0.22 <NA> <NA>
#> 2 nmr_s_ldl_p <NA> <NA> <NA> 7 =0.31
forestplot <- make_forest_plot(panels = list(resultsA),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.headings = c("Analysis A"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 0, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
col.heading.space = 1.5,
addtext = list(resultsA_extra),
right.space = unit(35, "mm"))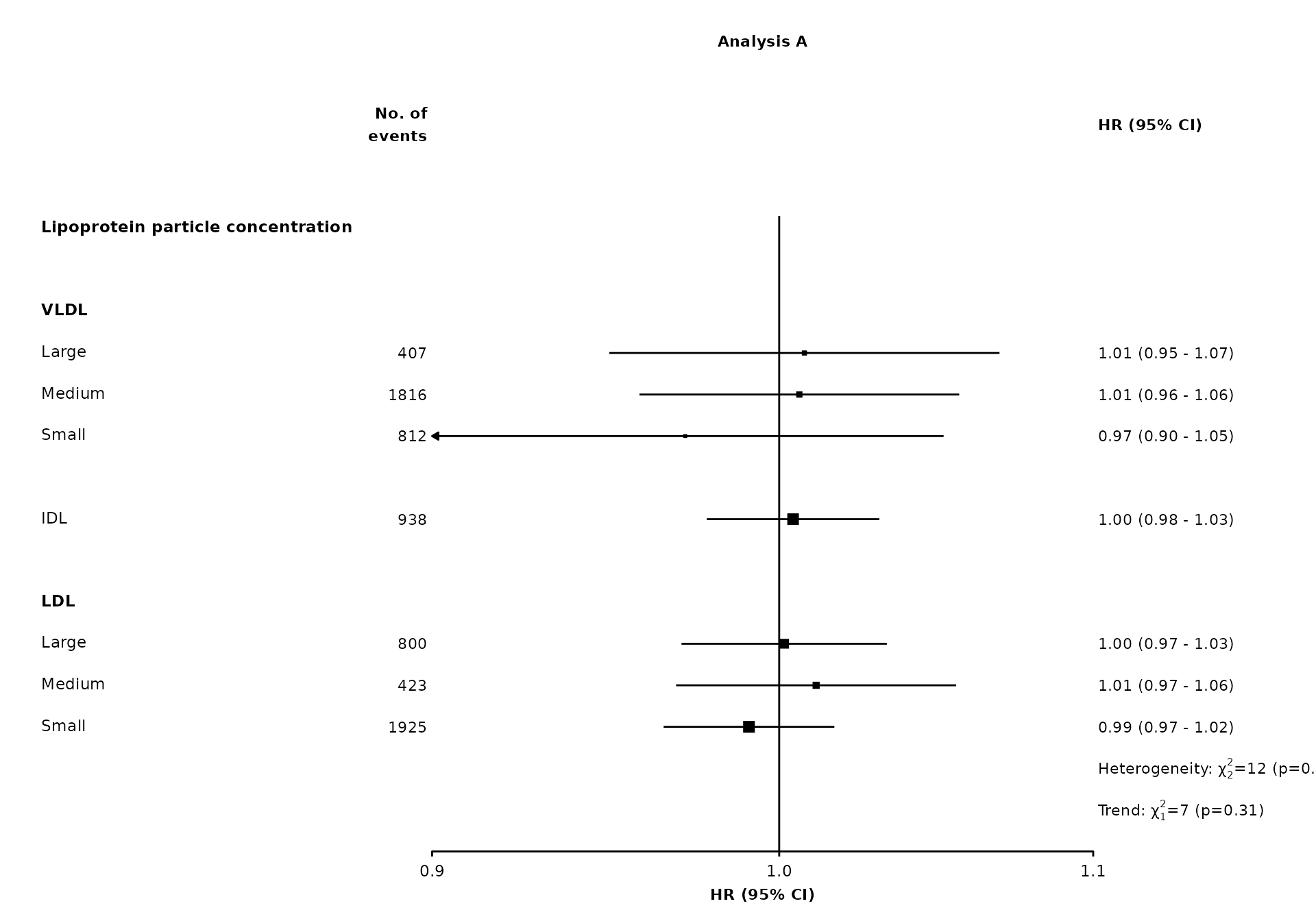
Different limits and ticks on each plot
make_forest_plot() uses ggplot facets to place forest plots side-by-side. Facets cannot easily have different scales (or limits or ticks) applied, so it’s not directly possible to have different limits and ticks on each forest plot.
However, one approach to work around this is to use make_forest_plot() for each plot you need, remove the labels from all but the first, then arrange them side-by-side. The gridExtra package can be used for this last step.
Step 1: Use make_forest_plot() for each plot.
forestplot1 <- make_forest_plot(panels = list(resultsA),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration",
"Triglycerides concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
col.heading.space = 1.5,
printplot = FALSE)
forestplot2 <- make_forest_plot(panels = list(resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration",
"Triglycerides concentration"),
exponentiate = TRUE,
panel.names = c("Analysis B"),
ci.delim = " - ",
xlim = c(0.8, 1.2),
xticks = c(0.8, 1, 1.2),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 3,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
col.heading.space = 1.5,
printplot = FALSE)Step 2: Remove the axis text for all but the first plot.
p1 <- forestplot1$plot
p2 <- forestplot2$plot +
theme(axis.text.y = element_blank())Step 3: Arrange the plots using gridExtra (there may be other packages that also work), using fix_panel() to set the same width for both panels. (Adjust the widths argument until you get a suitable layout.)
gridExtra::grid.arrange(fix_panel(p1, width = unit(20, "mm")),
fix_panel(p2, width = unit(20, "mm")),
nrow = 1,
widths = c(1, 0.5))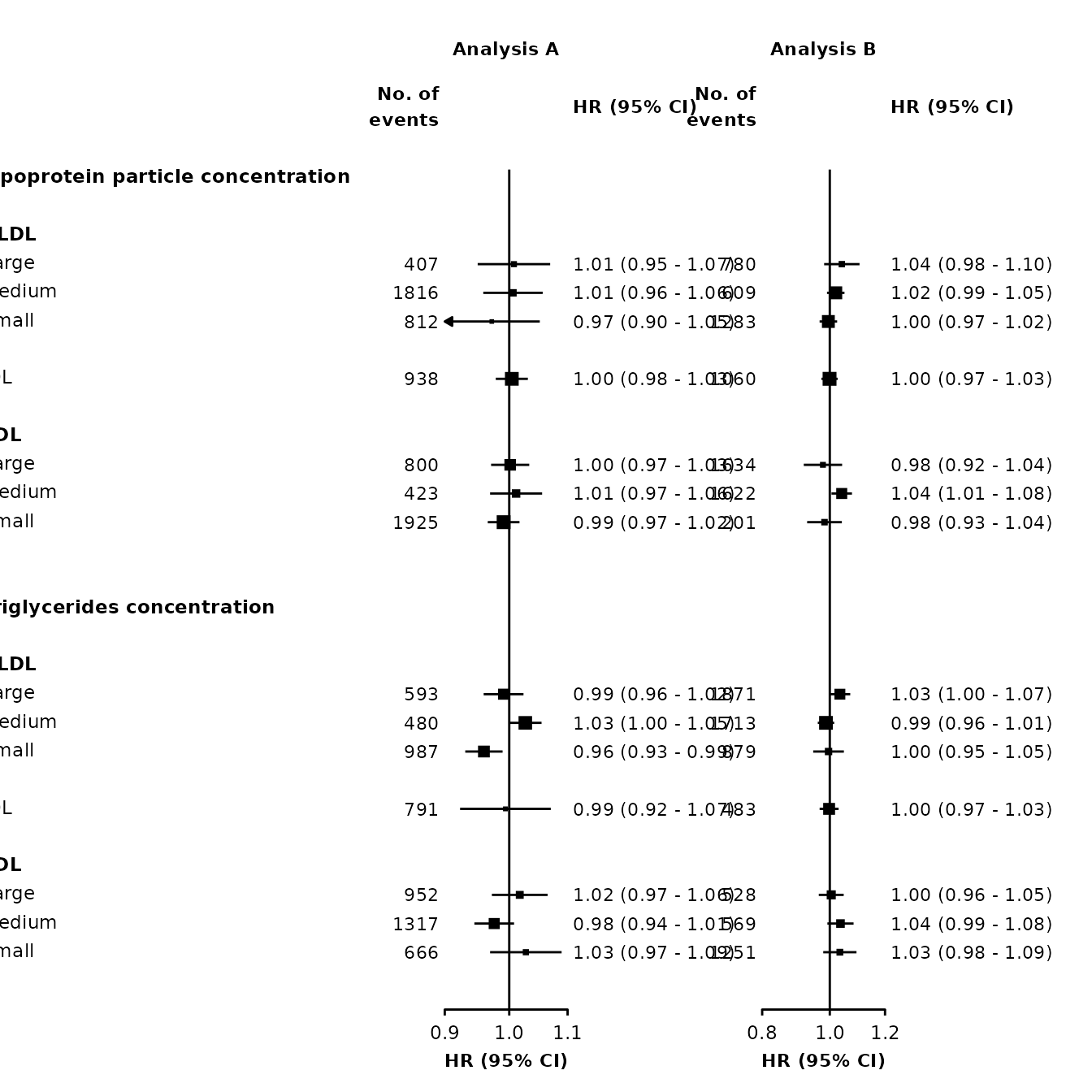
Note that if scalepoints = TRUE (and minse is not specified the same for each plot) then this scaling is on a plot-by-plot basis so box sizes are not comparable between plots. However, if different axis scales are used then confidence intervals are not comparable either so this may be not be a problem.
Changing narrow confidence interval lines
When making a forest plot, if a confidence interval line is narrower than the point representing the estimate we may wish to plot the line differently. In this package, this is possible by using the panel.width argument in the make_forest_plot() function, then using the fix_panel() function.
Changing the colour
To plot narrow confidence widths a different colour, set the panel.width argument of make_forest_plot() equal to the width each panel will be in the final drawn output. The cicolour argument should also be a character vector - the last element will be used for narrow confidence intervals.
Then use the fix_panel() function, where the first argument is the plot just created and the second is the same panel width. This function returns a gtable object, which can be plotted using grid::grid.draw (or plot()). The panels widths will be fixed.
Notes: This has been designed to work well for shape 15 (the default) and 22. Use a small value for stroke (< 0.5) to avoid a wide border which is not accounted for. The calculation of sizes may not be entirely accurate, so check the plot and change the plot.width argument as needed. Confidence intervals are assumed to be centred on the point estimate.
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 8,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
col.heading.space = 0,
# set panel width + CI colours
panel.width = 20,
cicolour = c("black", "white"),
printplot = FALSE)
plot <- fix_panel(forestplot$plot, width = unit(20, "mm"))
grid::grid.draw(plot)
The cicolour argument can be a vector of all names of colours, or all names of columns (which contain colour names).
resultsA$cicol1 <- "red"
resultsB$cicol1 <- "blue"
resultsA$cicol2 <- "white"
resultsB$cicol2 <- "white"
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 8,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
# set panel width + CI colours
panel.width = 20,
cicolour = c("cicol1", "cicol2"),
printplot = FALSE)
plot <- fix_panel(forestplot$plot, width = unit(20, "mm"))
grid::grid.draw(plot)
Plotting lines under or over points
As well as changing colour, we may also wish to change if confidence interval lines are plotted under or over the point estimates. This can be done using the plot.width argument and using a logical (or character) vector for the ciunder argument.
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 8,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
# set panel width + CI under or over
panel.width = 20,
shape = 22,
stroke = 0.5,
fill = "white",
ciunder = c(TRUE, FALSE),
printplot = FALSE)
plot <- fix_panel(forestplot$plot, width = unit(20, "mm"))
grid::grid.draw(plot)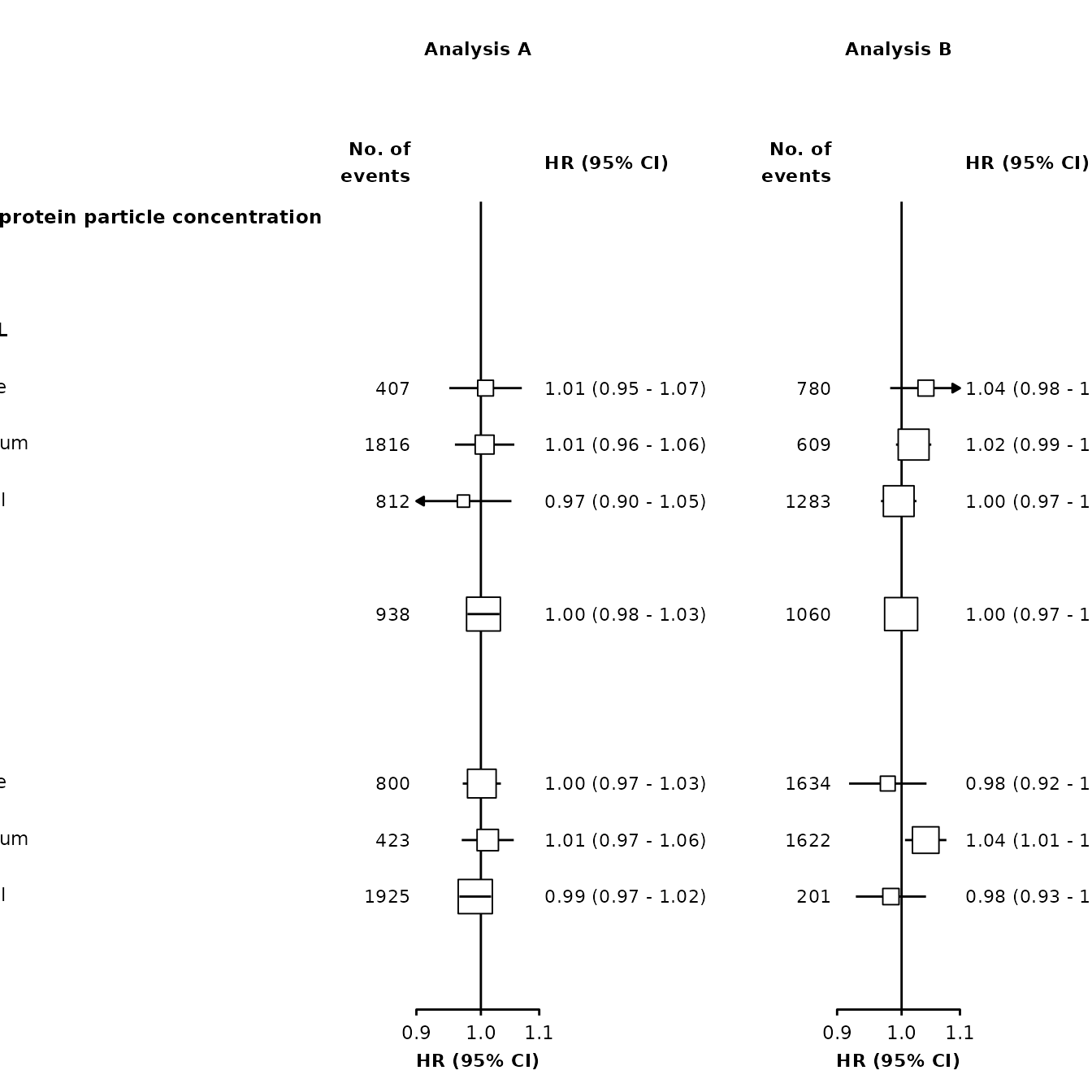
Changing colour and plotting of lines
resultsA <- dplyr::select(resultsA, -ciunder)
resultsA$cicol1 <- "black"
resultsA$cicol2 <- "white"
resultsA$shape <- 15
resultsB <- dplyr::select(resultsB, -ciunder)
resultsB$cicol1 <- "black"
resultsB$cicol2 <- "black"
resultsB$shape <- 22
forestplot <- make_forest_plot(panels = list(resultsA, resultsB),
col.key = "variable",
row.labels = ckbplotr_row_labels,
row.labels.levels = c("heading", "subheading", "label"),
rows = c("Lipoprotein particle concentration"),
exponentiate = TRUE,
panel.names = c("Analysis A", "Analysis B"),
ci.delim = " - ",
xlim = c(0.9, 1.1),
xticks = c(0.9, 1, 1.1),
blankrows = c(1, 1, 0, 1),
scalepoints = TRUE,
pointsize = 8,
col.left = c("n"),
col.left.heading = c("No. of\nevents"),
# set panel width + CI colours
panel.width = 20,
shape = "shape",
stroke = 0.5,
cicolour = c("cicol1", "cicol2"),
ciunder = c(TRUE, FALSE),
fill = "white",
printplot = FALSE)
plot <- fix_panel(forestplot$plot, width = unit(20, "mm"))
grid::grid.draw(plot)
Stroke
The stroke argument sets the stroke aesthetic for plotted shapes. See https://ggplot2.tidyverse.org/articles/ggplot2-specs.html for more details. The stroke size adds to total size of a shape, so unless stroke = 0 the scaling of size by inverse variance will be slightly inaccurate (but there are probably more important things to worry about).